GB 5009.5-2025 食品安全国家标准 食品中蛋白质的测定 
中华人民共和国国家标准
GB 5009.4—2025
食品安全国家标准
食品中蛋白质的测定
2025-03-16 发布 2025-09-16 实施
中华人民共和国国家卫生健康委员会
国 家 市 场 监 督 管 理 总 局发布
前 言
本标准代替GB 5009.5—2016《食品安全国家标准食品中蛋白质的测定》。
本标准与GB 5009.5—2016相比,主要变化如下:
—修改了标准适用范围;
—修改了第一法凯氏定氮法的取样量和标准滴定溶液浓度;
—增加了第二法标准溶液和显色剂的储存条件和时间;
—修改了第三法燃烧法的适用范围和检出限;
—增加了附录B燃烧法校正曲线;
—修改了分析结果表述和附录C蛋白质折算系数表;
—修改了精密度。
1 范围
本标准规定了食品中蛋白质的测定方法。
本标准适用于食品中蛋白质的测定。
第一法 凯氏定氮法
2 原理
食品中的蛋白质在催化加热条件下被分解,产生的氨与硫酸结合生成硫酸铵。碱化蒸馏使氨游离,用硼酸吸收后以硫酸或盐酸标准滴定溶液滴定,根据硫酸或盐酸的消耗量计算氮含量,再乘以折算系数,即为蛋白质的含量。
3 试剂和材料
除非另有说明,本方法所用试剂均为分析纯,水为GB/T 6682规定的三级水。
3.1试剂
3.1.1 硫酸铜(CuSO4 · 5H2 O) 。
3.1.2 硫酸钾(K2SO4 ) 。
3.1.3 氢氧化钠(NaOH) 。
3.1.4 硫酸(H2SO4 ) :≥98% 。
3.1.5 硼酸(H3BO3 ) 。
3.1.6 乙醇(C2H5 OH) :95% 。
3.1.7 甲基红(C15H15N3 O2 ) 。
3.1.8 溴甲酚绿(C21 H14Br4 O5 S) 。
3.1.9亚甲基蓝(C16 H18ClN3S · 3H2 O) 。
31.10 pH试纸 :测量范围 0~ 14。
3.2 试剂配制
3.2. 1 硼酸溶液(20g/L):称取20 g硼酸,加水溶解后并稀释至1 000 mL。
3.2.2 氢氧化钠溶液(400g/L):称取40 g氢氧化钠加水溶解后,冷却,并稀释至100 mL。
3.2.3 硫酸标准滴定溶液或盐酸标准滴定溶液[c(HCl)]0.10 mol/L。按照GB/T 601的要求配制和标定,或经国家认证并授予标准物质证书的滴定溶液标准物质。
3.2.4 硫酸标准滴定溶液或盐酸标准滴定溶液[c(HCl)]0.05 mol/L:用移液管吸取50 mL 0.10 mol/L硫酸标准滴定溶液或盐酸标准滴定溶液[c(HCl)]至容量瓶,用水稀释到100 mL,临用现配,必要时重新标定。或经国家认证并授予标准物质证书的滴定溶液标准物质。
3.2.5 甲基红指示剂(1g/L):称取0.1 g甲基红,溶于95%乙醇,稀释至100 mL,10℃~30℃,保存12个月。
3.2.6 亚甲基蓝指示剂(1g/L):称取0.1 g亚甲基蓝,溶于95%乙醇,稀释至100mL,10℃~30℃保存12个月。
3.2.7 溴甲酚绿指示剂(1g/L):称取0.1 g溴甲酚绿,溶于95%乙醇,稀释至100mL,10℃~30℃保存12个月。
3.2. 8 A混合指示液:2份甲基红乙醇溶液与1份亚甲基蓝乙醇溶液,混匀。10℃~30℃保存12个月。
3.2.9 B混合指示液:1份甲基红乙醇溶液与5份溴甲酚绿乙醇溶液,混匀。10℃~30℃保存12个月。
4 仪器和设备
4.1分析天平:感量为1 mg。
4.2 定氮蒸馏装置:如附录A所示。
4.3 移液管:10 mL、25 mL和50 mL。
4.4 定氮瓶:100 mL、250 mL和500 mL。
4.5 消化炉:≥420℃。
4.6 半自动凯氏定氮仪或全自动凯氏定氮仪 。
4.7 匀浆机 。
4.8 粉碎机。
5 分析步骤
5.1 凯氏定氮法
5.1.1 试样制备
液态样品摇匀;含有气体的蛋白饮料应采用超声波的方式脱气后再称量测定;基质均匀的半固态样品和粉状样品直接称量;其他样品需匀浆或粉碎均匀。粮食类样品至少要研磨200 g样品。颗粒、块状固体样品研磨后要充分混匀,使其完全通过0.9 mm(20目)孔径的筛子。产品标准中对样品处理有特殊要求的,按产品标准执行。制备好的试样应尽快测定。
5.1.2 水分测定
如试样结果以干基计算,应按照GB 5009.3或相应产品标准中规定的方法测定水分。
5.1.3 试样处理
称取固体试样0.2 g~2 g、半固体试样2 g~5 g,(精确至0.001g),液体试样10mL(g)~25mL(g)(相当于30mg~40mg氮),分别移入干燥的100mL、250mL或500mL定氮瓶中,加入0.4 g硫酸铜、6 g硫酸钾及20 mL硫酸,轻摇后于瓶口放一小漏斗,将瓶以45°角斜放置于有小孔的石棉网上。缓慢加热,待内容物全部碳化,泡沫完全停止后,加大火力,并保持瓶内液体微沸,至液体呈蓝绿色并澄清透明后,再继续加热0.5 h~1 h。取下定氮瓶冷却至室温,小心加入20 mL水,将内容物全部转移至100 mL容量瓶中,并用少量水洗定氮瓶内壁,洗液并入容量瓶中,再加水至刻度,混匀备用。同时进行空白试验。
5.1.4测定
按附录A装好定氮蒸馏装置,向水蒸气发生器内装水至2/3处,加入数粒玻璃珠,加甲基红数滴及数毫升硫酸,至溶液成红色,以保持水呈酸性,加热煮沸水蒸气发生器内的水并保持沸腾。
向接收瓶内加入10.0 mL硼酸溶液及3滴~4滴A混合指示剂或B混合指示剂,并使冷凝管的下端插入液面下,根据试样中氮含量,准确吸取2.0 mL~10.0 mL试样处理液由小玻杯注入反应室,以10 mL水洗涤小玻杯并使之流入反应室内,随后塞紧棒状玻塞。将10.0 mL氢氧化钠溶液倒入小玻杯,提起玻塞使其缓缓流入反应室,立即将玻塞盖紧,并水封。夹紧螺旋夹,开始蒸馏。蒸馏15 min后移动蒸馏液接收瓶,液面离开冷凝管下端,再蒸馏约1 min,至用pH试纸检测馏出液为中性。用少量水冲洗冷凝管下端外部,取下蒸馏液接收瓶。尽快以硫酸或盐酸标准滴定溶液滴定至终点,如用A混合指示液,终点颜色为紫红色;如用B混合指示液,终点颜色为浅灰红色。
5.2 全自动或半自动凯氏定氮仪法
称取固体试样0.2 g~2 g、半固体试样2 g~5 g,(精确至0.001g),液体试样10g(mL)~25g(mL)(相当于30mg~40mg氮),再加入0.4 g硫酸铜、6 g硫酸钾及20mL硫酸于消化炉进行消化。当消化炉温度达到420℃之后,继续消化至少1 h,此时消化管中的液体呈绿色透明状,于全自动或半自动凯氏定氮仪(使用前根据不同仪器优化分析参数,加入氢氧化钠溶液,盐酸或硫酸标准溶液以及含有混合指示剂A或B的硼酸溶液)进行试样检测。
当蛋白质含量≤1g/100g或1 g/100 mL时,建议使用0.05 mol/L的标准滴定液滴定,当蛋白质含量>1g/100g或1 g/100 mL时,建议使用0.10 mol/L的标准滴定液滴定。
注:当蛋白质含量过低且滴定体积小于1 mL或蛋白质含量过高导致滴定体积大于自动凯氏定氮仪滴定管体积时,可调整称样量以减小滴定误差。当样品脂肪含量过高或糖含量过高时,可调整称样量以减小或避免消化时出现爆沸、喷溅、溢流的情况。
6 分析结果的表述
试样中蛋白质的含量按式(1)进行计算 :
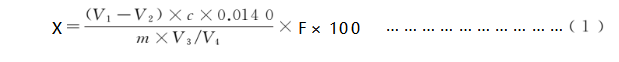
式中 :
X — 试样中蛋白质的含量 ,单位为克每百克或克每百毫升(g/100g或 g/100mL) ;
V1 — 试液消耗硫酸或盐酸标准滴定液的体积 ,单位为毫升(mL) ;
V2 — 试剂空白消耗硫酸或盐酸标准滴定液的体积 ,单位为毫升(mL) ;
c — 硫酸或盐酸标准滴定溶液浓度 ,单位为摩尔每升(mol/L) ;
0.0140—1.0mL硫酸mol/L或盐酸[c(½H2SO4)=1.000 mol/L]或烟酸[c(HCI)=1.000 mol/L]标准滴定溶液相当的氮的质量,单位为克每毫摩尔(g/mmoL);
m — 试样的质量 ,单位为克或毫升(g或 mL) ;
V3 — 吸取消化液的体积 ,单位为毫升(mL) ;
V4 — 消解溶液的定容体积 ,单位为毫升(mL) ;
F — 蛋白质折算系数(见附录 C) ;
100 — 由 g/g转化为 g/100 g 的换算系数 。
蛋白质含量≥1g/100 g或1 g/100 mL时,结果保留三位有效数字;蛋白质含量<1g/100 g或1 g/100 mL时,结果保留两位有效数字。
注1:分析结果以氮含量表述时,不需要乘蛋白质折算系数F。分析结果以蛋白质含量表述时,应同时报告蛋白质折算系数。
注2:当用半自动或全自动凯氏定氮仪全部转移消化液时,V3=V4。
注3:以干基计算试样中蛋白质的含量需根据试样的水分含量折算。
7 精密度
当样品中蛋白质含量≤10g/100g或10 g/100 mL,在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
当样品中蛋白质含量>10g/100g或10 g/100 mL,在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的5%。
8 其他
当用0.05 mol/L盐酸滴定液时,称样量为5.0 g或5.0 mL,本方法对氮的检出限为0.008 g/100 g或0.008g/100 mL。
第二法 分光光度法
9 原理
食品中的蛋白质在催化加热条件下被分解,分解产生的氨与硫酸结合生成硫酸铵,在pH 4.8的乙酸钠-乙酸缓冲溶液中与乙酰丙酮和甲醛反应生成黄色的3,5-二乙酰-2,6-二甲基-1,4-二氢化吡啶化合物。在波长400 nm下测定吸光度值,与标准曲线比较定量,结果乘以换算系数,即为蛋白质含量。
10 试剂和材料
除非另有规定,本方法中所用试剂均为分析纯,水为GB/T 6682规定的三级水。
10.1试剂
10.1.1 硫酸铜(CuSO4 · 5H2 O) 。
10.1.2 硫酸钾(K2SO4 ) 。
10.1.3 氢氧化钠(NaOH)。
10.1.4 对硝基苯酚(C6 H5NO3 ) 。
10.1.5 乙酸钠(CH3COONa· 3H2 O) 。
10.1.6 无水乙酸钠(CH3COONa) 。
10.1.7 硫酸(H2SO4 ) :98% 。
10.1.8 乙酸(CH3COOH) 。
10.1.9 甲醛(HCHO) :37% 。
10.1.10 乙酰丙酮(C5 H8 O2 ) 。
10.1.11 乙醇(C2 H5 OH) :95% 。
10.1.12 硫酸铵[(NH4 ) 2SO4] :优级纯 。
10.2 试剂的配制
10.2. 1 氢氧化钠溶液(300g/L):称取30 g氢氧化钠加水溶解后,放冷,并稀释至100 mL。
10.2.2 对硝基苯酚指示剂(1g/L):称取0.1 g对硝基苯酚指示剂溶于20mL 95%乙醇中,加水稀释至100 mL。
10.2.3 乙酸溶液(1 mol/L):量取5.8 mL乙酸,以水稀释至100 mL。
10.2.4 乙酸钠溶液(1 mol/L):量取41 g无水乙酸钠或68 g乙酸钠,溶于水并稀释至500 mL。
10.2.5 乙酸钠-乙酸缓冲溶液:量取60 mL乙酸钠溶液与40 mL乙酸溶液混合,该溶液pH为4.8。
10.2.6 显色剂:分别量取15 mL甲醛与7.8 mL乙酰丙酮,混合后以水稀释至100 mL,剧烈振摇混匀,4℃条件下于棕色瓶中可保存3 d。
10.3 标准品
硫酸铵标准储备溶液(以氮计)(1.0 g/L),或经国家认证并授予标准物质证书的标准品。
10.4 标准溶液配制
10.4.1 硫酸铵标准储备溶液(以氮计)(1.0 g/L):称取105℃干燥2 h的硫酸铵0.4720 g加水溶解后稀释至100 mL,混匀。此溶液每毫升相当于1.0 mg氮。4℃条件下可保存1个月。
10.4.2 硫酸铵标准工作溶液(0.10 mg/mL):准确吸取硫酸铵标准储备液(1.0 g/L)10.00 mL于100 mL容量瓶内,以水稀释至刻度,混匀。此溶液每毫升相当于0.1 mg氮。临用现配。
11 仪器和设备
11.1 分光光度计。
11.2 电热恒温水浴锅 :100 ℃ ±1 ℃ 。
11.3 pH 计 :精度 0. 01。
11.4 10 mL 具塞玻璃比色管 。
11.5 分析天平 :感量为 1 mg和 0. 1 mg。
11.6 定氮瓶 :100 mL、250 mL和 500 mL。
11.7 匀浆机 。
11.8 粉碎机。
12 分析步骤
12.1 试样制备同5. 1. 1。
12.2 水分测定同 5. 1. 2。
12.3 试样消解
称取固体试样0.1 g~0.5 g、半固体试样0.2 g~1 g(精确至0.01 g)、液体试样1 mL(g)~5 mL(g),移入干燥的100mL或250mL定氮瓶中,加入0.1 g硫酸铜、1 g硫酸钾及5 mL硫酸,轻摇后于瓶口放一小漏斗,将定氮瓶以45°角斜放置于有小孔的石棉网上。缓慢加热,待内容物全部炭化,泡沫完全停止后,加大火力,并保持瓶内液体微沸,至液体呈蓝绿色澄清透明后,再继续加热0.5 h~1 h。取下定氮瓶冷却至室温,小心加入20 mL水,将内容物全部转移至100 mL容量瓶中,并用少量水洗定氮瓶内壁,洗液并入容量瓶中,再加水至刻度,混匀备用。同时进行空白试验。
12.4 试样溶液的制备
量取2.0 mL~5.0 mL(精确至0.1 mL)试样或试剂空白消化液于50 mL或100 mL容量瓶内,加1滴~2滴对硝基苯酚指示剂溶液,摇匀后滴加氢氧化钠溶液中和至黄色,再滴加乙酸溶液至溶液无色,以水稀释至刻度,混匀。
12.5 标准曲线的制作
吸取0.00 mL、0.05 mL、0.10 mL、0.20 mL、0.40mL、0.60mL、0.80mL和1.00 mL硫酸铵标准使用溶液(相当于0.00μg、5.00μg、10.0μg、20.0μg、40.0μg、60.0μg、80.0μg和100.0μg氮),分别置于10 mL比色管中。加4.0 mL乙酸钠-乙酸缓冲溶液及4.0 mL显色剂,以水稀释至刻度,混匀。置于100℃水浴中加热15 min。取出用水冷却至室温后,移入1 cm比色杯内,以零管为参比,于波长400 nm处测量吸光度值,根据标准曲线各点吸光度值绘制标准曲线或计算线性回归方程。
12.6 试样测定
吸取0.50mL~2.00mL(相当于氮<100μg)试样溶液和同量的试剂空白溶液,分别置于10mL比色管中。加4.0 mL乙酸钠-乙酸缓冲溶液及4.0 mL显色剂,加水稀释至刻度,混匀。置于100℃水浴中加热15 min。取出用水冷却至室温后,移入1 cm比色杯内,以零管为参比,于波长400 nm处测量吸光度值,在标准曲线上查得待测液的浓度。
13 分析结果的表述
试样中蛋白质的含量按式(2)进行计算。
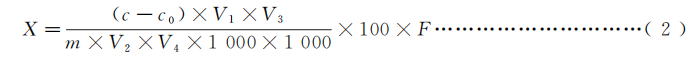
式中 :
X — 试样中蛋白质的含量 ,单位为克每百克或克每百毫升(g/100 g 或 g/100mL) ;
c — 试样测定液中氮的含量 ,单位为微克(μg) ;
c0 — 试剂空白测定液中氮的含量 ,单位为微克(μg) ;
V1 — 试样消化液定容体积 ,单位为毫升(mL) ;
V3 — 试样溶液总体积 ,单位为毫升(mL) ;
m — 试样质量 ,单位为克或毫升(g或 mL) ;
V2 — 制备试样溶液的消化液体积 ,单位为毫升(mL) ;
V4 — 测定用试样溶液体积 ,单位为毫升(mL) ;
1 000 — 由 μg转化为 mg的换算系数 ;
1 000 — 由 mg转化为 g 的换算系数 ;
100 — 由 g/g转化为 g/100 g 的换算系数 ;
F — 蛋白质折算系数(见附录 C) ;
蛋白质含量≥1g/100 g或1 g/100 mL时,结果保留三位有效数字;蛋白质含量<1g/100 g或1 g/100 mL时,结果保留两位有效数字。
注1:分析结果以氮含量表述时,不需要乘蛋白质折算系数F。分析结果以蛋白质含量表述时,应同时报告蛋白质折算系数。
注2:以干基计算试样中蛋白质的含量需根据试样的水分含量折算。
14 精密度
当样品中蛋白质含量≤10g/100g或10 g/100 mL,在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
当样品中蛋白质含量>10g/100g或10g/100 mL,在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的5%。
15 其他
称样量为5.0 g或5.0 mL时,本方法对氮的检出限为0.000 1 g/100g或0.000 1 g/100 mL。
第三法 燃烧法
16 原理
试样在900℃~1 200℃高温下燃烧,燃烧过程中产生混合气体,其中的碳、硫等干扰气体和盐类被吸收管吸收,氮氧化物被全部还原成氮气,形成的氮气气流通过热导检测器(TCD)进行检测。
17 试剂和材料
17.1 氧气(O2) :纯度 ≥99. 995% 。
17.2 载气(CO2 , He,Ar等) :纯度 ≥99. 995% 。
17.3 含氮标准物质 :天冬氨酸(C4 H7NO4 ) 、尿素(CH4N2 O) ,纯度 ≥99% 。 或经国家认证并授予标准物质证书的含氮标准物质 。
17.4 无氮铝箔纸 、锡囊 、无氮塑料囊或不锈钢坩埚 。
18 仪器和设备
18.1 氮/蛋白质分析仪:配有热导检测器 ,测定范围 0. 1 mg~ 200 mg氮含量 。具备校正功能 。
18.2 分析天平 :感量为 0. 1 mg。
18.3 匀浆机 。
18.4 粉碎机 。
19 试样制备
同 5. 1. 1。
20 水分测定
同 5. 1. 2。
21 测定步骤
21.1 仪器工作条件
21.1.1 燃烧温度
900 ℃ ~ 1 200 ℃ 。
21.1.2 通氧量
使用前应根据不同仪器自行检查最佳通氧量。通氧量应控制在样品燃烧后,保留2%~8%残氧量。
21.1.3 通氧时间
根据样品量的不同以及样品燃烧难易程度不同调节通氧气时间,以保证样品完全燃烧。
21.1.4 仪器校正
开机后待仪器稳定后,利用优化好的仪器参数测定氮含量略高于待测样品的标准物质,进行3次重复测定得到日常校正系数(f)。如日常校正系数大于1.1或小于0.9,或更换热导检测器后,应重新绘制校正曲线(见附录B),待日常校正系数结果在0.9~1.1范围内后,再进行样品测定。

式中 :
f — 日常校正系数 ;
c — 标准物质的理论氮含量 ,单位为克每百克(g/100g) ;
c1,c2,c3— 标准物质三次重复测定的氮含量 ,单位为克每百克(g/100g) 。
21.2 测定
根据仪器说明书要求称取0.1 g~1.0 g充分混匀的固体试样(精确至0.001 g),或移取0.1 mL(g)~1.0 mL(g)充分混匀的液体试样,用锡箔、锡囊包裹或称量于不锈钢坩埚中,放置于样品盘上。试样经过充分燃烧,测其含量。
22 分析结果的表述
试样中蛋白质的含量按式(4)进行计算 :
X =c × F …………………………( 4 )
式中 :
X — 试样中蛋白质的含量 ,单位为克每百克或克每百毫升(g/100g或 g/100mL) ;
c — 试样中氮的含量 ,单位为克每百克(g/100g) ;
F — 蛋白质折算系数(见附录 C) 。
蛋白质含量≥1g/100 g或1 g/100 mL时,结果保留三位有效数字;蛋白质含量<1g/100 g或1 g/100 mL时,结果保留两位有效数字。
注1:分析结果以氮含量表述时,不需要乘蛋白质折算系数F。分析结果以蛋白质含量表述时,应同时报告蛋白质折算系数。
注2:以干基计算试样中蛋白质的含量需根据试样的水分含量折算。
23 精密度
样品中蛋白质含量≤10g/100g或10 g/100 mL时,在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
样品中蛋白质含量>10g/100g或10 g/100 mL时,在重复条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的5%。
24 其他
称样量为0.2 g或0.2 mL时,本方法的检出限为0.50 g/100g或0.50 g/100 mL。

